
Research Article | DOI: https://doi.org/10.31579/2640-1053/017
*Corresponding Author: Mostafa A, Department of General Surgery, Faculty of Medicine, Egypt
Citation: Mostafa A, Charactor of JNK and P53 in Taxol-Induced Apoptotic Signaling in SKOV3 Human Ovarian Cancer Cell Proliferation. J Cancer Research and Cellular Therapeutics, Doi: 10.31579/2640-1053/017
Copyright: © 2017 Mostafa A . This is an open-access article distributed under the terms of The Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Received: 30 January 2017 | Accepted: 20 August 2017 | Published: 04 September 2017
Keywords: mitogen;phosphorylates;taxol
C-Jun N-terminal kinase (JNK) represents a group of mitogen-activated protein kinases (MAPKs) involved in many cellular responses including apoptosis. We have previously reported that taxol, a microtubule-interfering therapeutic agent widely used against various cancers, induces caspase-independent but apoptotic inducing factor (AIF)-dependent apoptosis in human ovarian cancer cell line SKOV3 cells. In the present study, we add to this report a detailed analysis of the taxol-induced apoptotic mechanisms in SKOV3 cells, particularly focusing on JNK and p53. In line with the previous report, we found that taxol induced caspase-independent apoptosis with concurrent activation of JNK, phosphorylation of Bcl-2, Bax translocation to the mitochondria, and AIF release from the mitochondria. Restoration of p53 functionality into SKOV3 cells, which are p53-null cells, by transfection of wild-type p53, however, induced caspase-dependent apoptosis in response to taxol treatment as evidenced by increasing PARP cleavage and the emergence of processed, active caspase-3 and -7. More to the point, treatment with a JNK inhibitor SP600125 blocked taxol-induced apoptotic cell death in both parental SKOV3 cells (p53-deficient) and p53-transfectant cells. Collectively, the aforementioned findings lend support to the view that taxol-induced apoptotic cell death in SKOV3 cells is executed by different mechanisms depending on the presence of p53 but commonly mediated by ASK1-JNK and/or -p38 axes.
C-Jun N-terminal kinase (JNK) represents a group of mitogen-activated protein kinases (MAPKs) involved in many cellular responses including apoptosis. We have previously reported that taxol, a microtubule-interfering therapeutic agent widely used against various cancers, induces caspase-independent but apoptotic inducing factor (AIF)-dependent apoptosis in human ovarian cancer cell line SKOV3 cells. In the present study, we add to this report a detailed analysis of the taxol-induced apoptotic mechanisms in SKOV3 cells, particularly focusing on JNK and p53. In line with the previous report, we found that taxol induced caspase-independent apoptosis with concurrent activation of JNK, phosphorylation of Bcl-2, Bax translocation to the mitochondria, and AIF release from the mitochondria. Restoration of p53 functionality into SKOV3 cells, which are p53-null cells, by transfection of wild-type p53, however, induced caspase-dependent apoptosis in response to taxol treatment as evidenced by increasing PARP cleavage and the emergence of processed, active caspase-3 and -7. More to the point, treatment with a JNK inhibitor SP600125 blocked taxol-induced apoptotic cell death in both parental SKOV3 cells (p53-deficient) and p53-transfectant cells. Collectively, the aforementioned findings lend support to the view that taxol-induced apoptotic cell death in SKOV3 cells is executed by different mechanisms depending on the presence of p53 but commonly mediated by ASK1-JNK and/or -p38 axes.
Paclitaxel or commonly called taxol is an anti-neoplastic agent specifically targeting microtubules, promoting microtubule depolymerization and thereby alters microtubule dynamics [1,2]. Such microtubule damage leads to the activation of mitotic spindle checkpoint whereby a transient arrest of the cell cycle in the mitosis may occur [2]. After mitotic arrest, cells can enter into apoptosis or exit from mitosis without dividing. The taxol-induced apoptotic signaling appears to be transmitted mainly via the mitochondria-dependent pathway which involves the release of cytochrome c and subsequent activation of caspase 9 and 3 [3]. c-jun N-terminal kinase (JNK) activation has also been observed in cells treated with microtubule targeting agents including paclitaxel [4,5].
JNK represents a group of mitogen-activated protein kinases (MAPKs) involved in many cellular responses including apoptosis [6]. A wide range of biological outcomes have been attributed to JNK activation, including enhanced proliferation or survival, after an exposure to stress, but the majority of published evidence supports the JNK’s role in conveying apoptotic signals when cells are exposed to DNA damage or anticancer therapies. A variety of JNK substrates including ATF-2, c-Jun, Bcl-2, Bcl-XL, p21Cip1/Waf1, and p53 have been identified, providing insight into the ultimate effects of activated JNK within varying cellular contexts [7]. JNK is activated by sequential phosphorylation through a MAP kinase module (MAPKKK and MAPKK). Once activated, JNK phosphorylates and regulates the activity of transcription factors such as the AP-1 family member and c-Jun [6]. It has also been unequivocally documented that JNK is necessary and sufficient for the activation of an intrinsic cell death pathway, and that pro-apoptotic members of the Bcl-2 family, BAX or Bak, are recruited for the execution of JNK-dependent apoptosis [8].
Microtubule-interfering agents seem to activate JNK through a signal transduction involving apoptosis signal-regulating kinase-1 (ASK1) [9,10]. ASK1, a 160 kDa serine/threonine protein kinase, is a member of the mitogen-activated protein kinase kinase kinase (MAPKKK) family and activates both p38 MAPK and JNK by directly phosphorylating and/or via activating SEK1 (also known as MKK4) [11-15]. The ASK1-mediated JNK activation causes the phosphorylation of Bcl-2, leading to a reduction of its anti-apoptotic activity [16]. However, the detailed molecular mechanism that links mitochondria-dependent apoptosis and ASK1-p38/JNK activation remain largely unknown [17].
p53 tumor suppressor protein is a short-lived transcription factor that serves as a key player in the cellular response to a variety of extra- and intracellular insults such as DNA damage, oncogenic activation, and microtubule disruption [18,19]. p53 exerts its function mainly through the transcriptional activation of target genes such as CDK inhibitor and p21waf1/Cip1 for arresting the cell cycle, and the pro-apoptotic protein, Bax, for inducing apoptosis [20]. Similar to other stresses, microtubule disruption results in an increase in p53 phosphorylation at multiple sites in a drug- and cell-specific manner with a resultant accumulation of transcriptionally active proteins [21,22]. In fact, accumulating evidence suggest that p53 is a major mediator in DNA damage-induced apoptosis [23].
Previously, we reported a caspase-independent but AIF-dependent apoptosis in association with taxol treatment in SKOV3 human ovarian cancer cells. Extending this finding, in this study, we have demonstrated that the phosphorylation of JNK is a key molecular event in the taxol-induced apoptotic signal transduction in SKOV3 cells. Furthermore, we have also provided evidence that Bcl-2 is bound to, thus phosphorylated by activated JNK, and BAX is subsequently translocated into the mitochondria, leading to AIF release from the mitochondria. However, when p53 is restored into SKOV3 cells, the taxol-induced apoptotic signaling is shifted from a caspase-independent to a caspase-dependent signaling, raising an intriguing possibility of conferring on p53 a new role in the apoptotic cell death.
Materials and Methods
Antibodies and reagents
Paclitaxel was purchased from Sigma-Aldrich (St. Louse, MO). SP600125, a JNK inhibitor, was from Merck (Darmstadt, Germany). RPMI 1640 medium and penicillin/streptomycin solution were from Gibco® Invitrogen (Grand Island, NY). Heat-inactivated fetal bovine serum (FBS) was from WelGene (Seoul, S. Korea). TransIT-LT1® was from Mirus Bio (Madison, WI). 5,5’-6,6’-tetrachloro-1,1’-3,3’-tetraethylbenzimidazolyl-carbocyanine iodide (JC-1) and streptoavidin-conjugated Alexa Flour 488 dye (FITC) were from Molecular Probe (Eugene, OR). Mouse monoclonal anti-Bcl-2, anti-JNK, anti-p-JNK, rabbit polyclonal anti-AIF, anti-p-c-Jun antibodies were from Cell Signaling Technology (Beverly, MA). Anti-p-ERK, anti-ERK, horse radish peroxidase (HRP)-conjugated anti-mouse and anti-rabbit secondary antibodies were from Santa Cruz Biotech (Santa Cruz, CA). Biotin conjugated goat anti-rabbit and mouse secondary antibodies were from ZYMED (San Francisco, CA). Protease and phosphatase inhibitor cocktail containing PMSF, pepstain, leupeptin, and aprotinin was from Calbiochem (La Jolla, CA). Protein A/G-plus agarose® was from Santa Cruz Biotech. Polyvinylidene difluoride (PVDF) membrane was from Amersham (Piscataway NJ). Enhanced chemiluminescence reagents were from Pierce (Rockford, IL). TACS™ MTT cell proliferation assay kit was from R&D systems (Minneapolis, MN). Bio-Rad protein assay reagent was from Bio-Rad (Hercules, CA). Fluorescent mounting medium was from DAKO (Carpinteria, CA). Mitochondria isolation kit was from Pierce (Rockland, IL). The pCMVp53 vector that harbors the wild type p53 cDNA was from Clontech (Madison, WI). Other chemicals unless otherwise specified were from Sigma-Aldrich.
Cell culture and transfection of p53
Human ovarian SKOV3 carcinoma cells were purchased from American Tissue Culture Collection (ATCC, HTB-77) and maintained at 37°C in RPMI 1640 medium supplemented with 10
Statistical analysis
One-way ANOVA was utilized for the statistical analysis of experimental results by using the SPSS 10.0 software. P < 0> was taken as significance.
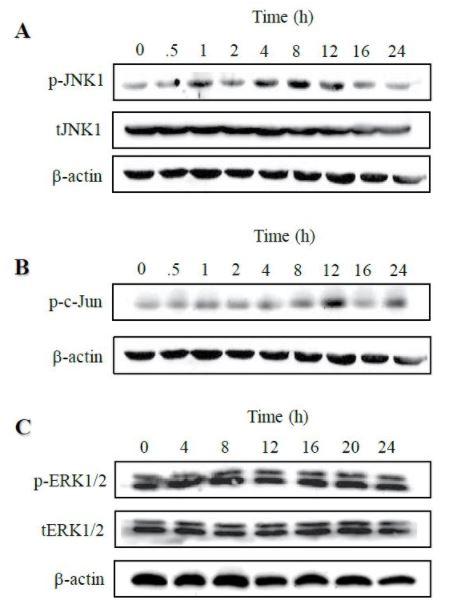
Activation of JNK by taxol
Despite various therapeutic outcomes of taxol in different types of cancer, less is known about the signaling pathways whereby taxol conveys its cytotoxic and apoptotic effects on cancer cells. JNK activation in response to chemotherapy treatment has been suggested to correspond to cell sensitivity to chemotherapy [25]. To this regard, we sought to test if taxol activated JNK-mediated signaling pathways, characteristically including JNK and c-Jun in SKOV3 cells. JNK activity was assessed by immunoblotting with phospho-specific anti-p-JNK1 antibodies recognizing major phosphorylation sites on Thr-183 and Tyr-185. First, we examined the time course of JNK1 activation following taxol treatment. SKOV3 cells were treated with 100 nM paclitaxel for varying amounts of time (0 - 24 hrs), and cellular levels of p-JNK1 and its substrate p-c-Jun were measured by immunoblotting. As illustrated in Figure 1, the phosphorylation of JNK1 and c-Jun were time-dependent. The phosphorylation of JNK1 was stimulated by taxol treatment, appearing as early as at 1 hr after taxol treatment and steadily increasing with a maximum activation observed at 8 hr, and then declining to the basal level at 24 hrs following drug treatment. Consistent with this observation, the emergence of p-c-Jun, a substrate of active JNK, lagged behind that of p-JNK1, reaching the peak at 12 hr before declining. On the other hand, unlike a recent documentation that taxol may induce a transient ERK activation [26], paclitaxel at 100 nM did not influence ERK activation in SKOV3 cells as evidenced by little changes in phospho-ERK1/2 immunoreactivity over the 24 hr-period, strongly suggesting that the ERK-mediated MAP kinase pathway is not activated by taxol.
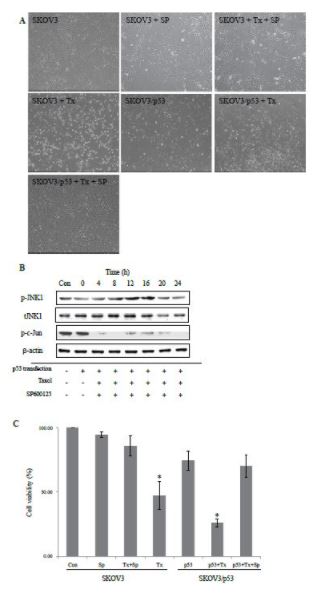
SP600125 suppresses apoptotic cell death in SKOV3 cells
To further ensure that the taxol-induced JNK activation is associated with apoptotic cell death in SKOV3 cells, we used a JNK inhibitor, SP600125, to see if its treatment deterred the taxol-induced cell death. Photomicrographs in Figure 2a show morphologies of apoptotic cells following paclitaxel and/or SP600125 treatment. Exposure to 100 nM paclitaxel for 24 hrs clearly induced cell death as evidenced by the emergence of round-shaped and buoyant cells seen in Figure 2a(D), which was almost completely inhibited by co-treatment of 25 mM SP600125 (Figure 2a(C)). We then restored the wild-type p53 into SKOV3 cells, which are p53-null, and examined if p53 alleviated the taxol-induced cell death. Contrary to our expectation, the introduction of p53 did not alleviate the taxol-induced cell death nor inhibit SP600125 ability to deter the taxol-induced cell death (Figure 2a(E)-(G)). These results were further confirmed by measuring JNK activity under parallel conditions. Immunoblotting analyses showed that SP600125 affected the phosphorylation of JNK and c-Jun but in an opposite direction. The phosphorylation of c-Jun was barely detectable in the presence of 25 mM SP600125 while that of p-JNK1 was not significantly affected by SP600125 at the same concentration up to 8 hr treatment (Figure 2b). Seen in Figure 2(c) are the extents of apoptotic cell death caused by taxol in the presence of SP600125 under different physiological contexts in terms of p53 as quantified by MTT. 100 nM taxol treatment increased cell death substantially in comparison with control (p < 0>), which were apparently reversed almost to the control level by 25 mM SP600125 co-treatment. A similar trend was observed with p53-transfected SKOV3 cells (SKOV3/p53 cells), suggesting that p53 may not antagonize cell death caused by taxol but rather may sensitize the taxol-induced cell death, raising a possibility that p53 activation may represent an active involvement in executing the taxol-induced cell death.
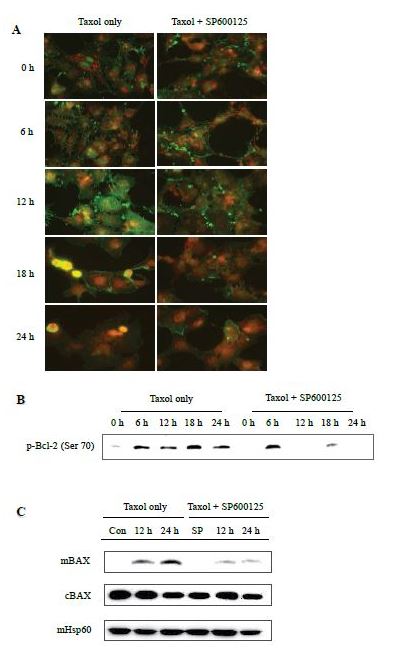
Based on previous documentation that the p-JNK-mediated apoptosis depends on the recruitment and activation of Bcl-2 [16,29], we next measured the binding of p-JNK1 with p-Bcl-2 by the immunoprecipitation assay. Whole-cell lysates were prepared from cells that were pretreated with 100 nM paclitaxel and/or 25 mM SP600125 for 0-24 hrs and subjected to immunoprecipitation by antibodies recognizing p-JNK1. The resulting precipitates were then immunoblotted with anti-p-Bcl-2 antibodies. Taxol increased the amount of p-Bcl-2 co-precipitated with p-JNK1 in a time-dependent manner, which remained up-regulated for up to 24 hrs but was abrogated by SP600125 co-treatment (Figure 3b).
Proapoptotic Bax is suggested to normally exist in the cytoplasm [30,31], and ample evidence suggests that Bax has both active and inactive conformations that principally determine its subcellular distribution [32]. In response to apoptotic stimuli, Bax changes its subcellular localization and dimerization pattern in such a way that homo-dimerized Bax is transformed into a membrane-bound form and thus translocated to the mitochondria, leading to the initiation of mitochondria-dependent apoptosis [33-35]. To determine whether taxol increase Bax translocation onto the mitochondria, the mitochondria were isolated and then analyzed by immunoblotting analysis with antibodies recognizing Bax. Taxol increased mitochondrial Bax in a time-dependent manner, which was also subjected to attenuation by SP600125 treatment while cytoplasmic Bax remained unchanged following drug treatments (Figure 3c).
SP600125 attenuates the taxol-induced mitochondrial depolarization and AIF release
JC-1 is a dual fluorescent membrane potential-sensitive dye that fluoresces at 590 nm (red fluorescence) in mitochondrial hypolarization and at 510 nm (green fluorescence) in mitochondrial depolarization (loss of mitochondrial transmembrane potential Dym). Mitochondrial depolarization is known to indicate an early event of apoptosis, which leads to increased mitochondrial membrane permeability and release of proapoptotic factors such as AIF into the cytosol [36]. We investigated whether Dym loss occurred following taxol treatment in a SP600125-sensitive manner. As illustrated in Figure 4a, treatment with taxol induced mitochondrial depolarization as evidenced by higher green fluorescence than untreated cells, and this transition was clearly attenuated by SP600125 co-treatment. Also demonstrated in Figure 4, that the taxol-induced release of AIF from the mitochondria into the nucleus was greatly attenuated by the addition of SP600125 (Figure 4b).
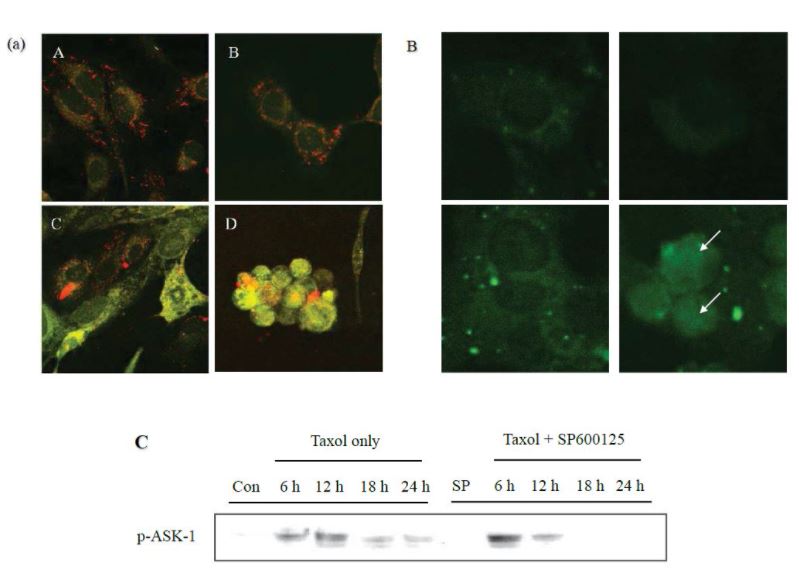
To explore an upstream regulator of JNK activity, which might account for the early response to taxol treatment, we examined the phosphorylation status of ASK1 following taxol treatment. Figure 4(c) shows that phosphorylated ASK1 began to appear at as early as 6 hrs and reached the peak at 12 hrs following taxol treatment. However, unlike p-JNK1 and p-c-Jun, SP600125 was not likely to attenuate the phosphorylation of ASK1, strongly implying that ASK1 may be an upstream regulator of JNK1, at least, in the taxol-induced apoptotic signaling pathway in SKOV3 cells.
Caspase-dependent apoptosis is induced when p53 is restored in SKOV3 cells
It has been well established either loss of wild type p53 function, gain of oncogenic function, or the ability to activate p53 inappropriately severely compromises the capacity for controlled cellular proliferation and growth [37]. In addition, despite the fact that both p53 and JNK have previously been shown to mediate cellular responses to the actions of a microtubule-interfering agent [10], their interrelationships have been far from clear. To gain further insight into the mechanism regulating the p53-mediated JNK activation, wild-type p53 was reintroduced by transfection into SKOV3 cells, which are deficient of endogenous p53. Then, we characterized the taxol-induced apoptotic signaling in SKOV3/p53 cells. First, the expression level of p53 was measured by immunoblotting. As shown in Figure 5a, the expression level of p53 was significantly elevated compared to parental cells (control), and in addition, p53 expression level was not susceptible to either taxol and/or SP500125 treatment. The presence of cleaved poly-ADP-ribose polymerase (PARP) is one of the most frequently used diagnostic tools for the detection of caspase-dependent apoptosis in many cell types. The cleavage of PARP into two fragments of 89 and 24 kDa has been considered indicative of functional caspase activation [38]. Figure 5b revealed an increase in cleaved forms of PARP when SKOV3 cells were exposed to taxol. In addition, the induction of cleaved PARP by taxol was further augmented in SKOV3/p53 cells, implying that the induction of caspase-dependent apoptosis is p53-dependent. Intriguingly, this induction was attenuated by SP500125 treatment. More consistently, the emergence of processed thus active caspase-3 and -7 was also induced by 100 nM taxol treatment when wild type p53 was restored in SKOV3 cells in a SP600125-sensitive manner. These data clearly suggest that p53 may play a pivotal role in the activation of caspase-3 and -7 and the induction of caspase-dependent apoptotic cell death in SKOV3 cells in response to taxol.

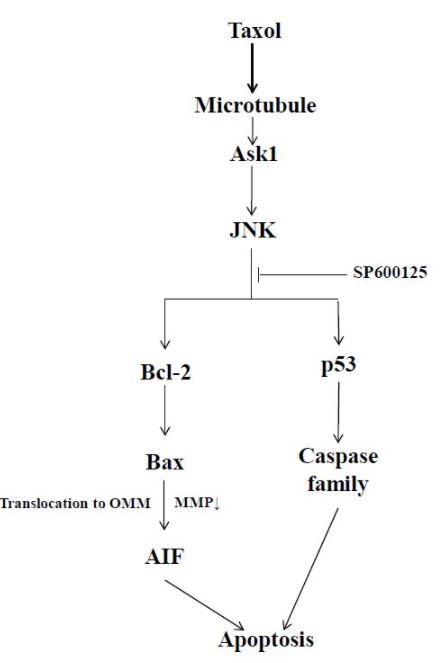
In the present study, we have provided evidence that supports the involvement of JNK in the taxol-induced apoptotic cell death in SKOV3 cells. The emerging view from the present studies is that apoptotic cell death is executed via the following signaling pathway in SKOV3 cells in response to taxol treatment: Taxol induces the activation of ASK1, which, in turn, increases the phosphorylation of JNK. Activated JNK binds to and thus activates mitochondrial Bcl-2 concomitant with mitochondrial Bax translocation. Translocated Bax may cause mitochondrial depolarization, which facilitates the release of AIF from the mitochondria into the nucleus. AIF in the nucleus eventually leads to apoptotic cell death via a process, generally termed ‘caspase-independent apoptosis’ as suggested elsewhere [36]. Further supporting our reasoning is the finding that treatment of SKOV3 cells with SP600125, a JNK inhibitor, attenuates apoptotic cell death with concurrent inhibition of signaling events that must occur downstream of JNK, such as Bcl-2 phosphorylation, mitochondrial Bax translocation, mitochondrial depolarization, and mitochondrial AIF release. Additionally, the restoration of p53 in SKOV3 cells can restore ‘caspase-dependent apoptotic cell death’ as evidenced by the emergence of cleaved form of PARP and caspase-3 and -7 activation upon receiving taxol treatment. We interpret this to mean that SKOV3 cells may activate two different apoptotic signaling pathways in response to microtubule-interfering agents, e.g., taxol: caspase-independent and caspase-dependent signaling pathway depending on the presence of p53. Therefore, p53 may play a switchboard role in the taxol-induced apoptotic cell death in such a way that without p53, like parental SKOV3 cells, caspase-independent but mitochondria-dependent apoptotic signaling is prevalent while the introduction of p53 causes a signaling shift to caspase-dependent apoptosis. Furthermore, the finding that SP60012 is effective in blocking cell deaths in both p53-null and p53-containing SKOV3 cells raises a possibility that JNK may be a key molecule in both caspase-independent and caspase-dependent apoptotic signaling pathways in SKOV3 cells.
Of note, recent documentation suggests
Since biochemical studies led to the identification and purification of JNK as a ‘p54 microtubule-associated protein kinase’ [39], JNK was found to bind the NH2-terminal activation domain of c-Jun [40,41] and thus phosphorylate c-Jun on ser-63 and ser-73 [42]. JNK is activated by treatment of cells with cytokines (e.g., TNF and IL-1) and by exposure of cells to many forms of environmental stress (e.g., osmotic stress, redox stress, and radiation) [43]. Several MAPKKK have been reported to activate the JNK signaling pathway, including members of the apoptosis signal-regulating kinase (ASK) group. Apoptosis signal-regulating kinase1 (ASK1) is characterized as a MAPK kinase kinase. Overexpression of ASK1 induces apoptosis in mink lung epithelial cells, and ASK1 is activated in cells treated with tumor necrosis factor-?, suggesting a role of ASK1 in stress-, cytokine-, and microtubule-interfering agents (MIAs)-induced apoptosis [9, 44]. However, the exact mechanism whereby JNK is activated in the stress-response is unclear. Alternatively, JNK activation may represent a protective response that is initiated by an exposure to stress. In fact, the JNK pathway has been implicated in both apoptosis and survival signaling [43]. On the other hand, JNK activation has been reported to induce cytochrome c release [45], but the mechanism is unclear. Potential targets of JNK that may regulate cytochrome c release may include members of Bcl-2 group of apoptotic regulatory proteins [16]. Phosphorylation of Bcl-2 and/or Bcl-XL inhibits the anti-apoptotic function of these proteins. However, there are some problems with this potential mechanism. First, one study indicates that Bcl-2 phosphorylation on the same sites may be anti-apoptotic rather than pro-apoptotic [46]. The second problem is that other stimuli cause a marked Bcl-2 phosphorylation without activating JNK. Finally, the other apoptotic mechanism is independent of Bcl-2 phosphorylation and caspase activation. This reasoning may demonstrate that anti-apoptotic proteins Bcl-2 and Bcl-XL is not physiological substrates of JNK, and rather, JNK could be targeted to Bcl-2 or Bcl-XL under specialized circumstances in the presence of an adaptor molecule that mediates the interaction. Although it is established that JNK contributes to some apoptotic response, it is not clear that apoptosis represents the only functional consequence of JNK activation and indeed JNK-dependent apoptotic signaling pathway can be blocked by activation of survival signaling pathways [47], including NF-?B, Akt/PKB and ERK.
Similarly, a significant question that remains unanswered concerns the molecular mechanism that can account for the function of JNK in apoptotic signal transduction in response to taxol. Our present data shows that taxol is not implicated in ERK phosphorylation. Indeed, our previous reports have shown that paclitaxel and selenium compounds both caused apoptosis in SKOV3 through a caspase-independent pathway [36,48]. JNK activation has been reported to induce Bcl-2 phosphorylation [2,16]. Likewise, our data provide evidence that JNK signaling pathway directly regulates the apoptotic mechanism in a mitochondria-mediated fashion and activated JNK is directly responsible for the phosphorylation of Bcl-2 and Bax translocation onto the mitochondria. These findings are consistent with previous results of Nechushtan et al. [33], who clearly demonstrated in electron microscopy that Bax translocation to the outer mitochondrial membrane was the first step in Bax activation, and that the mitochondria-associated Bax oligomer was the biologically active pro-apoptotic structure. The acquisition of a membrane embedded conformation, which is associated with oligomerization of Bax, facilitates the release of mitochondrial proteins such as cytochorme c or AIF [32].
Other potential targets of pro-apoptotic signaling activated by JNK could be tumor suppressor protein p53. When JNK is activated in cells exposed to stress, JNK phosphorylates p53, thereby inhibiting ubiquitin-mediated degradation, and stabilizing p53 protein [49]. However, many other studies also pointed out that although JNK may contribute to the regulation of p53 stability, p53 does not appear to be required for JNK-induced apoptosis [50]. Our findings strongly suggest the involvement of p53 in apoptotic cell death in response to taxol treatment. Moreover, we provide ample evidence for a novel functional link between p53 and caspase-dependent cell death as outlined in Figure 6. At this point, it is not sure whether this novel linkage is cell-type specific or more or less ubiquitous irrespective of cell contexts.
Of note, it is worth mentioning that a number of recent papers demonstrated evidence that supports that wild type SKOV3 cells executes taxol-introduced cell death in a caspase-3-dependent manner [51-53]. It is well documented that the activation of caspase 3, a main executor protease in apoptosis, is again under heavy influence of various upstream signals coming from both extrinsic (death ligand) and intrinsic (mitochondrial) pathways; some are positive while others are negative regulators depending on the physiological context. Therefore, it is not surprising that other studies support the apoptotic cell death in wild type SKOV3 cells after taxol treatment is executed in a caspase-3-dependent manner. The physiological context of wild type SKOV3 cells could be various depending on, for instance, culture conditions, cell passage numbers (cell history), whether or not transfected with a foreign gene, and etc. What we saw in this study was a pair-wise comparison between wild type SKOV3 cells and P53-transfected SKOV3 cells in terms of the emergence of cleaved form of caspase 3. And our results clearly saw a difference that the cleaved form of caspase 3 only came from P53-transfectants in response to taxol but negligible in wild type SKOV3 cells. Also supporting this argument, we have published similar results previously [36]. So, it is remained to be seen whether wild type SKOV3 cells recruit caspase 3 when they commit the taxol-introduced cell death.
In conclusion, our study suggests that the p53-mediated apoptosis may play a significant role in apoptotic cell death of many cancer cells, including ovarian cancer, in response to a variety of anticancer chemotherapeutics. Deletion or mutation(s) of wild type p53 could lead to the elevation of the threshold for anticancer drugs or the attenuation of their efficacies against cancers due to the abrogation of the p53-mediated apoptosis. For this reason, the p53-mediated apoptosis deserves a more stringent investigation to gain further insight into a new strategy for promoting cancer cell death and leading to efficient cancer prevention.
Dear Editorial Team, Clinical Medical Reviews and Reports. My experience with the journal was highly positive. The peer-review process was rigorous, constructive, and completed in a timely manner. The reviewers provided valuable comments that helped improve the quality and clarity of our manuscript. The editorial office was professional, responsive, and supportive throughout all stages of the publication process. Communication was clear and efficient, and any questions were addressed promptly. Overall, I found the journal to maintain high scientific standards and an excellent publication workflow. I would be pleased to consider submitting future work to this journal. Best wishes from, Elena Popa.

It was my pleasure to submit my testimonial concerning the Reviewer Board of our Scientific Journal “Brain and Neurological Disorders”. The Reviewers focused on some modifications and their contribution was helpful. The ladies of our Editorial Office were also supported my efforts. It was my honor to have such a co-operation and I am looking forward for more collaboration.

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Thank you for the speedy and efficient peer review process. I appreciate the fact that your peer reviewers do not take months to respond like with some other journals. I would also like to thank the editorial office for responding quickly to my questions. It is an excellent journal. I plan to submit more manuscripts in the future. Best wishes from, Robert W. McGee

Dear Grace Pierce, Editorial Coordinator of Journal of Clinical Research and Reports, Working with you and your team on our recent publication in JCRR has been a truly wonderful and enjoyable experience. The responses were prompt, and the reviewers were patient, constructive, and highly professional. One reviewer in particular gave me the feeling that a professor was carefully reading and commenting on my coursework, which was deeply touching. The entire process was straightforward and hassle‑free, with no tedious online forms to complete. I highly recommend this journal. Best wishes from, DR Aibing Rao, Head of R&D

I Appreciate the Opportunity to Share my Experience with the Journal of Clinical Research and Reports. The peer review process was timely and constructive, and the feedback provided helped improve the quality of our manuscript. The editorial office was professional, responsive, and supportive throughout the process, ensuring smooth communication and efficient handling of the submission. Overall, it was a positive experience collaborating with your team.

Dear Mercy Grace, Editorial Coordinator of Obstetrics Gynecology and Reproductive Sciences, We would like to express our gratitude for your help at all stages of publishing and editing the article. The editors of the magazine answer all the necessary questions and help at every stage. We will definitely continue to cooperate and publish other works in the Obstetrics Gynecology and Reproductive Sciences! Best wishes from, Alla Konstantinovna Politova,
